Workflow Overview
These documents describe a standardized Nextflow workflow for processing DIA mass spectrometry data to quantify peptides and proteins. The source code for the workflow can be found at: https://github.com/mriffle/nf-skyline-dia-ms.
This workflow supports three search engines: DIA-NN, Encyclopedia, and Cascadia for performing de novo searches. Each search engine works as a drop-in replacement for the other, supporting all the same pre- and post-analysis steps. In all cases, the workflow supports converting RAW files, integrating with PanoramaWeb (ProteomeXchange) and Proteomic Data Commons, and will generate a Skyline document suitable for visualization and analysis in Skyline.
Cascadia workflow:
The workflow will perform de novo identification of peptides using user-supplied DIA RAW (or mzML) files. The workflow will generate a Skyline document where users may visualize the de novo results and export integrated peak areas for the identified peptides.
DIA-NN workflow:
The workflow will quantify peptides and proteins using user-supplied DIA RAW (or mzML) files, FASTA file, and spectral library (optional). If the user does not specify a spectral library, DIA-NN will be run in “library-free” mode, where it will create its own library using AI. Finally the workflow will generate a Skyline document using the quantified peptides and proteins.
EncyclopeDIA workflow:
This workflow is summarized in the following article:
Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, Villén J, MacCoss MJ. Nat Commun. 2018 Dec 3;9(1):5128. (https://pubmed.ncbi.nlm.nih.gov/30510204/)
The workflow will quantify peptides and proteins using user-supplied DIA RAW (or mzML) files, FASTA file, and spectral library. If the experimental design includes generation of a chromatogram library using narrow window DIA data, the workflow will first generate the chromatogram library (Figure 1A) and use that as input to the next phase (Figure 1B) to quantify peptides and proteins. If the experimental design does not include this, the user-supplied spectral library is used as input for quantifying peptides and proteins. Finally the workflow will generate a Skyline document using the quantified peptides and proteins.
The workflow is summarized graphically as:
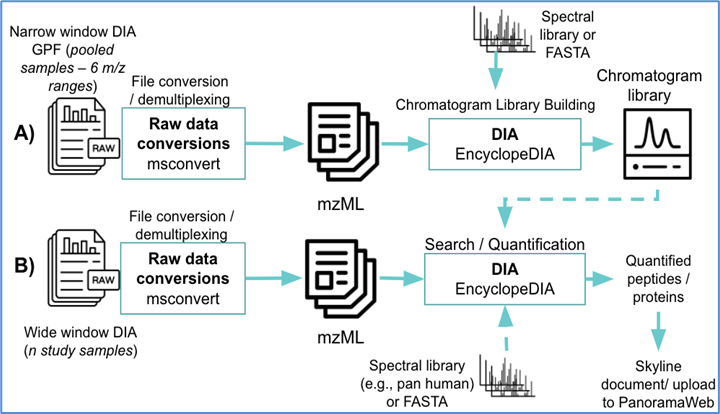
Figure 1. An overview of the computational pipeline implemented by this workflow. (A) the optional generation of a chromatogram library that can be fed into part (B) for peptide and protein quantification using DIA. If part (A) is not run, a user-supplied spectral library or chromatogram library may be used for quantification in part (B).
How to Run
This workflow uses the Nextflow standardized workflow platform. The Nextflow platform emphasizes ease of use, workflow portability, and containerization of the individual steps. To run this workflow, you do not need to install any of the software components of the workflow. There is no need to worry about installing necessary software libraries, version incompatibilities, or compiling or installing complex and fickle software.
To run the workflow you need only install Nextflow, which is relatively simple. To run the individual steps of the workflow on your own computer, you will need to install Docker. After these are installed, you will need to edit the pipeline configuration file to supply the locations of your data and execute a simple Nextflow command, such as:
nextflow run -resume -r main mriffle/nf-skyline-dia-ms -c pipeline.config
The entire workflow will be run automatically, downloading Docker images as necessary, and the results output to
the results directory. See How to Install the Workflow for more details on how to install Nextflow and Docker. See
How to Run the Workflow for more details on how to run the workflow. And see Output & Results for more details on how to
retrieve the results.
Workflow Components
The workflow is made up of the following software components, each may be run multiple times for different tasks.
PanoramaWeb (https://panoramaweb.org/home/project-begin.view)
Users may optionally use WebDAV URLs as locations for input data files in PanoramaWeb. The workflow will automatically download files as necessary.
msconvert (https://proteowizard.sourceforge.io/)
If users supply RAW files as input, they will be converted to mzML using msconvert.
EncyclopeDIA (http://www.searlelab.org/software/encyclopedia/index.html)
EncyclopeDIA is used in three parts of the pipeline:
If the user supplies a BLIB spectral library, EncyclopeDIA will be used to convert that to a DLIB.
EncyclopeDIA is used to search narrow window DIA data and generate a chromatogram library.
EncyclopeDIA is used to quantify peptides and proteins.
Skyline (https://skyline.ms/project/home/begin.view)
Skyline is run to import raw scan data EncyclopeDIA results into a Skyline template file.